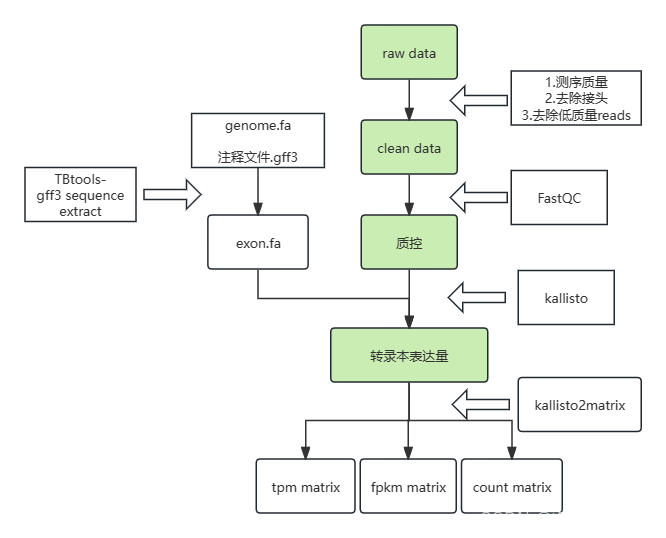
转录组上游-windows使用kallisto-从cleandata到表达矩阵
30人参与 • 2024-08-02 • Windows
由于我linux系统崩了,于是我开始探索再windows环境完成rna-seq分析,实际情况是windows完全够用(如果内存足够),不然还是选择用服务器分析。
网上对于kallisto的使用教程并不详细,也主要集中在linux系统,于是我想分享一下我使用kallisto的经验。这是我的分析流程,大家可以参考一下。
1.安装kallisto
kallisto是一个免费的转录组拼接软件,在linux和windows-cmd里都可以运行,使得你的row data被拼接为可以进行下游操作的read count这样的matrix。以下是下载安装的官网:
https://pachterlab.github.io/kallisto/download
下载到你能找得到的位置(很关键,因为等下要转到这个工作目录)下载后在文件夹里看到这样就成功下载安装了:
2.在cmd中运行kallisto
我们使用windows的命令行来操作,win+r打开运行,输入cmd进入命令行窗口;
然后我们要cd我们的工作目录到kallisto的目录:
>cd c:\users\huzhuocheng\desktop\毕业设计\转录组\转录组上游\kallisto\kallisto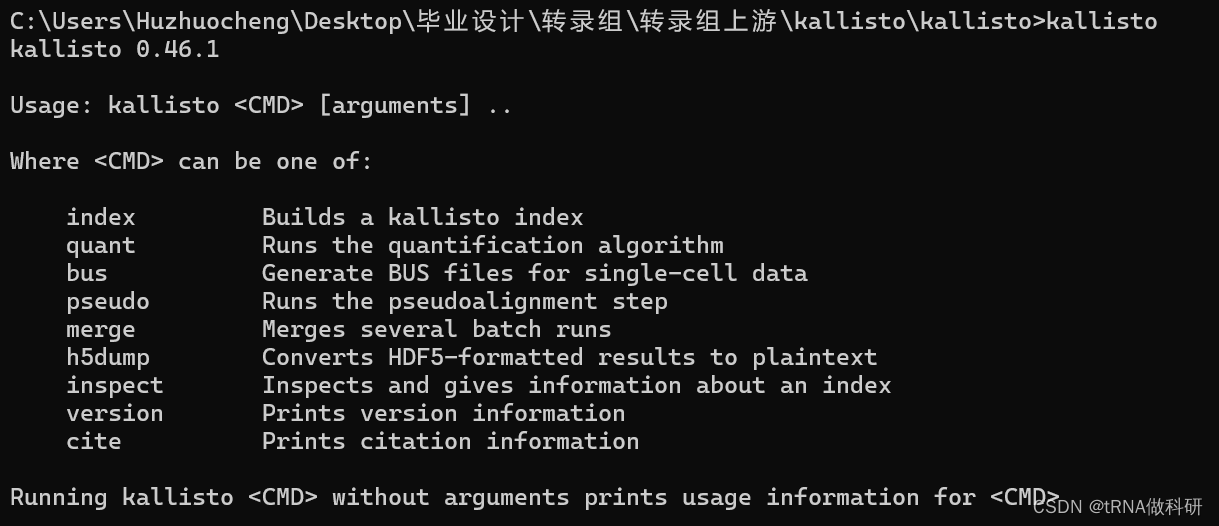
这样就欧克了,我们已经在kallisti的工作目录里了;
3.获取cds或者exon建立index
现在的转录组分析大多是有参转录组的分析了,不过kallisto也只能做有参的,因此要先建立索引。我们不能用全基因组做参考,这样切出来会一坨一坨的,因此我们需要先获得cds序列或者exon序列再来建立index,这里我推荐使用tbtools获取,tbtools转换需要两个文件:
(1)全基因组.fa(2)基因组注释文件.gff3
(没有这些文件的话可以在ncbi上找,我也会出教程,但是如果是冷门模式生物,比如我的萨氏海鞘,就可以问问实验室的大老板应该是有基因组文件的)
这样获得的cds.fa 或者 exon.fa就可以用来建立index,当然具体用cds还是exon可以自己选,我做出来发现cds表达量高一些,cds(coding sequence)和exon(外显子)都是基因结构中的重要组成部分。cds指的是一段特定的dna序列,它包含了用于编码蛋白质的所有信息。具体来说,cds是从基因的起始密码子(通常是atg)开始,一直到终止密码子(可以是taa、tag或tga)结束的一段序列。在这段序列中,每一个三联体密码子对应着蛋白质中的一个氨基酸,因此cds是基因表达成蛋白质的直接模板。
另一方面,exon则是指真核生物基因中的编码序列,这些序列在rna剪接过程中会被保留下来,并最终构成成熟的mrna分子。外显子本身可以跨越几个不同的dna片段,并且在剪接过程中会被连接在一起,形成连续的mrna序列。
总的来说,cds关注的是dna序列中负责编码蛋白质的部分,而exon则是从dna层面描述哪些序列会在最终的mrna中被表达出来。
4.kallisto建立index
在刚刚的cmd里输入kallisto index会跳出指令教程:
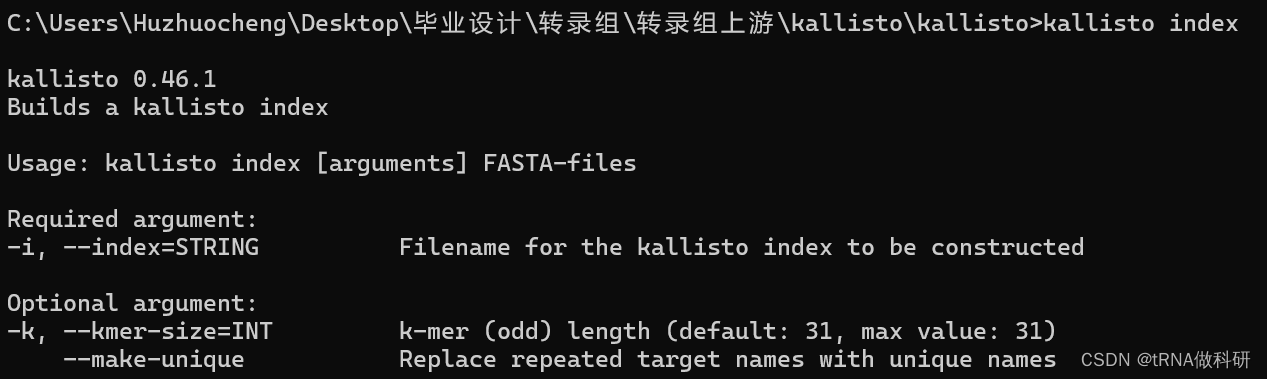
用法(usage):
kallisto index [arguments] fasta-files
#example
c:\users\huzhuocheng\desktop\毕业设计\转录组\转录组上游\kallisto\kallisto>kallisto index -i d:\cds.idx d:\cds.fakallisto index -i 你的输出位置 你的cds文件位置
必需的参数(required argument):
-i, --index=string: 你需要指定一个输出文件的名称,kallisto将用这个文件名来保存生成的索引。
可选的参数(optional argument):
-k, --kmer-size=int: 这个参数允许你设置k-mer的长度。k-mer是在构建索引过程中使用的短序列片段,通常长度为奇数个碱基对。默认值是31,最大值也是31。--make-unique: 如果你的输入文件中包含重复的目标名称(例如不同的转录本具有相同的名称),你可以使用此选项让kallisto生成唯一的名称以区分它们。
看到这样的反馈就是成功了:
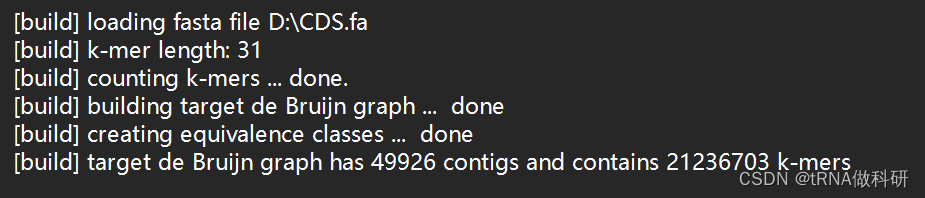
5.kallisto进行双端转录组测序数据定量
kallisto可以对双端和单端的测序数据进行比对,我这里以双端的为例。
在cmd里输入kallisto quant后,会出现比对指令表:
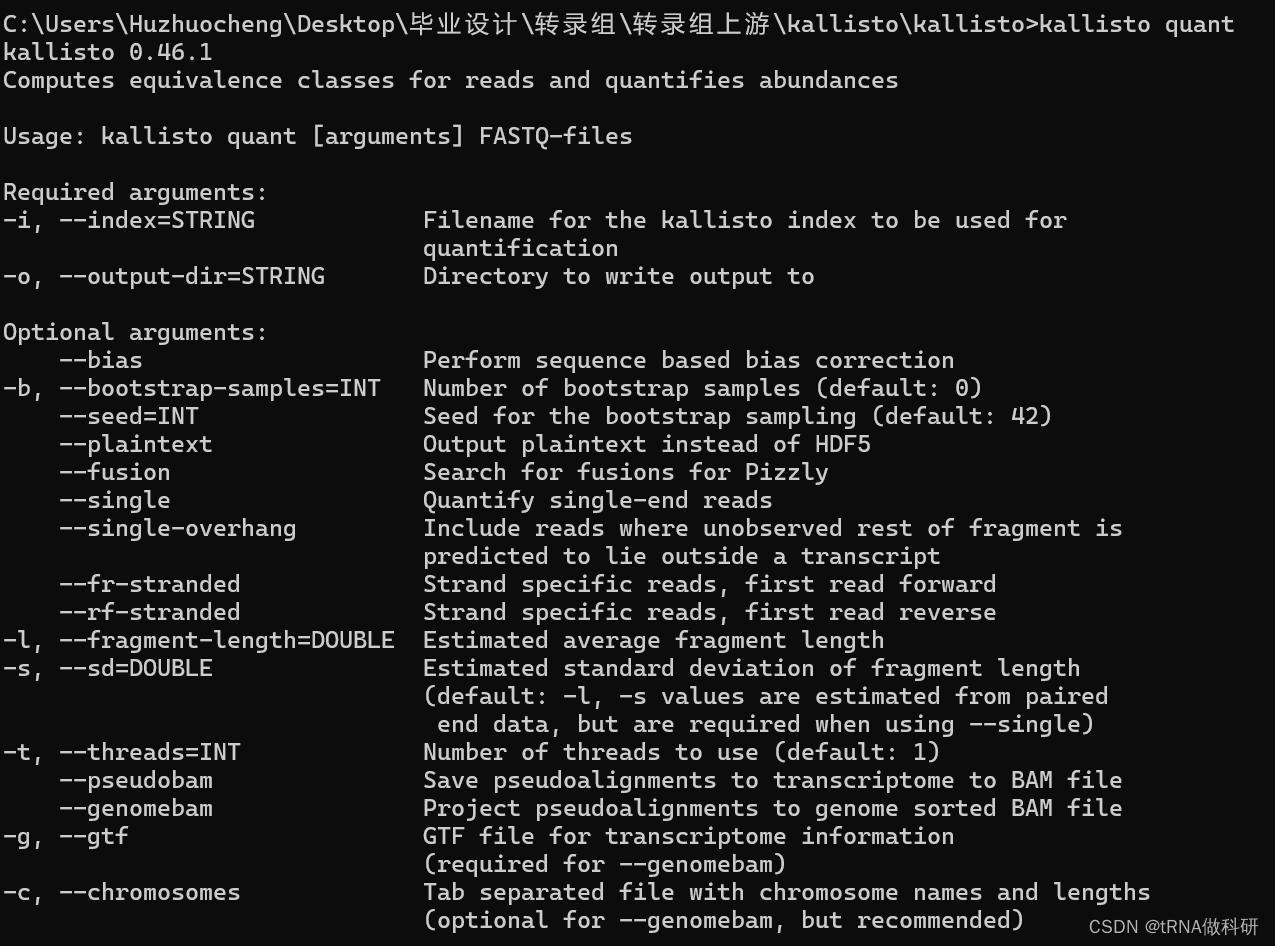
required arguments: 这一部分列出了运行kallisto时必须要提供的参数:
-i, --index=string: 指定用于定量分析的kallisto索引文件的名称。
-o, --output-dir=string: 指定输出结果的目录路径。
optional arguments: 接下来列出的是一些可选参数,用户可以根据需要选择是否提供这些参数:
--bias: 执行基于序列的偏差校正。
-b, --bootstrap-samples=int: 设置bootstrap采样的数量,默认为0。
--seed=int: 设置bootstrap采样的种子值,默认为42。
--plaintext: 以纯文本形式输出结果,而不是hdf5格式。
--fusion: 为pizzly搜索融合事件。
--single: 对单端读取数据进行定量。
--single-overhang: 包括预测其未观察到的片段剩余部分位于转录本之外的读取。
--fr-stranded: 针对正向链特异性读取(第一读取为正向)。
--rf-stranded: 针对反向链特异性读取(第一读取为反向)。
-l, --fragment-length=double: 估计的平均片段长度。
-s, --sd=double: 估计的片段长度的标准差,默认情况下是从配对末端数据中估计出来的,但在使用--single时需要明确指定。
-t, --threads=int: 要使用的线程数,默认为1。
--pseudobam: 将伪比对到转录本的记录保存为bam文件。
--genomebam: 将伪比对投影到基因组并生成排序后的bam文件。
-g, --gtf: 包含转录本信息的gtf文件,对于--genomebam选项是必需的。
-c, --chromosomes: 染色体名称和长度的制表符分隔文件,对于--genomebam选项是可选的,但推荐使用
大部分指令用不到,可以直接看我的示例:
c:\users\huzhuocheng\desktop\毕业设计\转录组\转录组上游\kallisto\kallisto>kallisto quant -i d:\cds.idx -o d:\second\hsl-1-1 d:\trans\clean\unknown_bt072-002t0001_good_1.fq d:\trans\clean\unknown_bt072-002t0001_good_2.fq
########格式
>kallisto quant -i index位置 -o 输出文件位置 第一个fq文件位置 第二个fq文件位置
看到这样就是运行成功了,可以去输出文件下看一下输出的文件。

6.使用kallisto2matrix转为read counts文件
刚刚得到的abundance文件还是不能直接分析,我们是想得到表达量的matrix文件。
下载地址是:github - yukaiquan/kallisto2matrix: kallisto/salmon result to matrix (tpm,count,fpkm)
我把它安装到了kallisto同一个目录,也是可以在cmd进行运行的
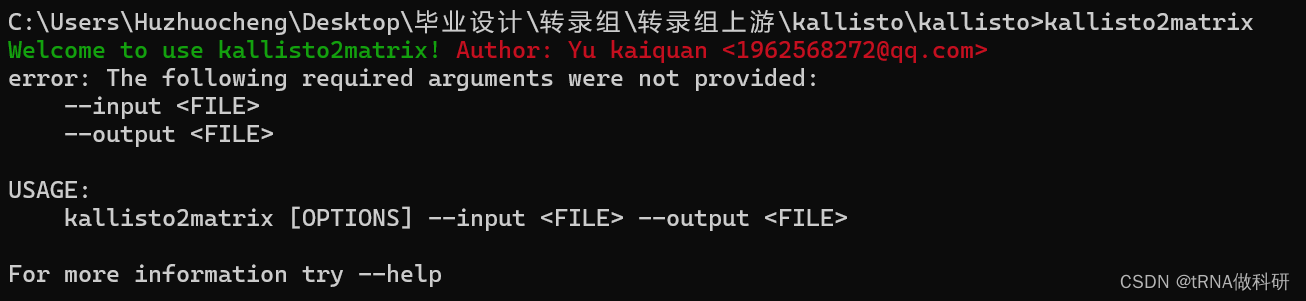
这样就安装成功了。
我们需要在这个工作目录下新建一个txt文件,里面放着我们刚刚得到的多个样本的abundance文件,然后运行指令就可以输出了。
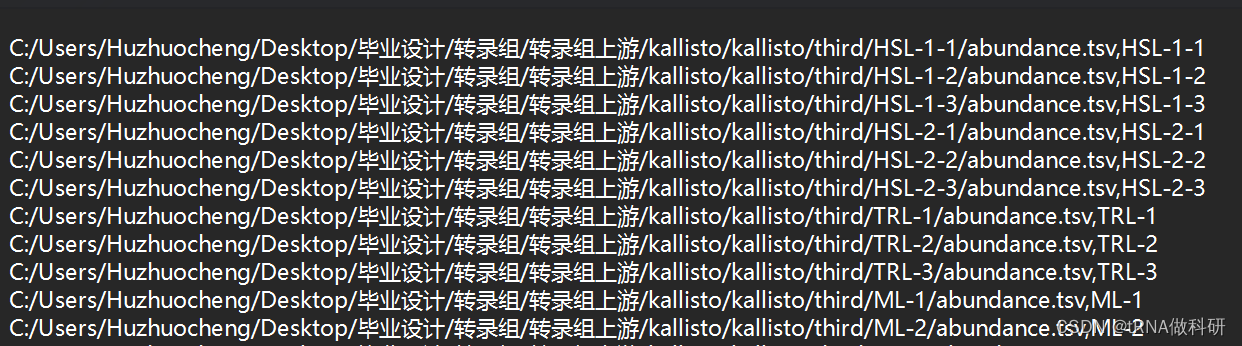
c:\users\huzhuocheng\desktop\毕业设计\转录组\转录组上游\kallisto\kallisto>kallisto2matrix.exe -i c:\users\huzhuocheng\desktop\毕业设计\转录组\转录组上游\kallisto\kallisto\example.txt -o test输出得到的是count/tpm/fpkm三个矩阵
1. counts矩阵代表原始的读数(read counts),它记录了每个样本中每条基因序列被测序仪检测到的次数。这是一个绝对数值,反映了测序的深度和基因的长度。由于这些因素可能导致不同样本或基因之间的直接比较存在偏差,因此通常需要通过其他形式的标准化来处理。
2. fpkm(fragments per kilobase of transcript per million mapped reads)矩阵是一种标准化后的表达量度量单位,旨在解决由基因长度和测序深度带来的问题。fpkm首先考虑了基因的长度,然后根据每个样本中映射到基因组的读取总数进行调整。具体来说,fpkm通过将每个基因的片段数(fragments)除以该基因的长度(以千碱基为单位),再乘以一百万(因为测序深度可能达到数百万级别),从而使得不同长度的基因和不同测序深度的样本之间可以进行比较。
3. tpm(transcripts per million)矩阵也是用来标准化基因表达量的一种方法,它与fpkm的主要区别在于调整顺序的不同。tpm先对每个样本的所有基因的读数进行归一化,然后再除以基因的长度。这种计算方法有助于在不同的样本之间进行更准确的比较,尤其是在跨样本分析时。tpm的一个显著特点是,所有样本中所有基因的tpm值加起来等于同一个常数(通常是1百万),这使得tpm非常适合于描述样本中基因表达的相对比例。
总结来说,这三个矩阵代表了从原始数据到标准化表达量的不同阶段。counts矩阵提供了最基础的读数信息,而fpkm和tpm则是对这些数据进行标准化处理后得到的表达量度量,它们帮助我们在不同样本间进行更有意义的比较和分析。
结语
以上就是从转录组raw data到表达量数据的全过程,后续的分析我也会出教程,有什么问题可以评论交流。谢谢大家!
赞 (0)
您想发表意见!!点此发布评论



发表评论